| . |
PRION? WHAZAT?
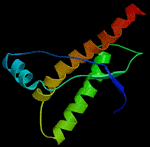 You'll also hear TSEs referred to as "Prion Diseases" (pronounced PREE on). TSEs are mostly likely, but not conclusively proved to be caused by "self-replicating" proteins or prions. Instead of staying in their normal shape, they fold into spirals.These spiraled prions can attach to other proteins of the same type that are normal and cause them to change shape. This produces a chain reaction causing the disease to grow. Image: Human Prions You'll also hear TSEs referred to as "Prion Diseases" (pronounced PREE on). TSEs are mostly likely, but not conclusively proved to be caused by "self-replicating" proteins or prions. Instead of staying in their normal shape, they fold into spirals.These spiraled prions can attach to other proteins of the same type that are normal and cause them to change shape. This produces a chain reaction causing the disease to grow. Image: Human Prions
In 1997, Dr. Stanley Prusiner won the Nobel Prize for his discovery
of prions - a term he coined in 1982. Back then, Prusiner took
a LOT of flack for this hypothesis since "bugs" were thought to
be transmitted only by bacteria or viruses, not proteins.
Though the prion theory has yet to be embraced 100%, it is the
most widely accepted explanation of these diseases.
A ROSE BY ANY OTHER NAME
There are lots of words tossed around when people talk about these
diseases and sometimes they are (mistakenly) lumped together as
"mad cow". All forms of this disease come under the broad heading
of Transmissible Spongiform Encephalopathy (TSE) whether it's found in animals or humans.
TSEs In Animals
The best known and most widely discussed is
BSE - (Bovine Spongiform Encephalopathy) which occurs in cattle. Other
variations include:
CWD - (Chronic Wasting Disease) a similar version found in elk and
deer
TME - Transmissible Mink Encephalopathy
FSE - Feline Spongiform Encephalopathy (domestic cat, puma, cheetah,
ocelot, tiger)
Scrapie or OSE - Ovine Spongiform Encephalopathy (sheep and goats)
Spongiform Encephalopathy of Exotic Ruminants (nyala, gemsbok, Arabian oryx, eland, kudu, scimitar-horned oryx, ankole, and bison)
TSEs In People
In humans, TSEs cover six diseases:
CJD - Creutzfeldt-Jakob Disease which can take up to 30 years to
incubate was first documented in 1920.
nvCJD or NV-CJD - new variety of CJD first seen in 1994-95; takes much less time
to manifest.
Alpers Syndrome - this is prion disease in infants.
|
Kuru - (meaning "tremble")

Soup's On!
|
Kuru results from cannibalism and was practiced by the Fore tribe
in the New Guinea Highlands. In this gruesome custom, the dead
were honored through ritualistic cannibalism. Children (and friends)
typically dined on the brains of their deceased parents. Ground
up into a pale gray soup, they heated and ate the brain. This
organ carries the highest concentration of the disease.
Other accounts say the entire body was cut up into parts, and
the men reserved the best (and least diseased) part, muscle, for
themselves. The remaining parts of the body, including brain,
pancreas, liver, kidney and intestines, were eaten by the women
and the children.6 This explains the higher death rate in women from kuru than men.
|
Many of the Fore got Kuru though sometimes it took up to 30 years to show up. Kids who participated in cannibalism developed the illness sooner than their older siblings or peers who ate this yummy entree.7 Since 1957 about 2,600 cases have been identified, but less than
10 cases existed by 19958. Today this disease is nearly wiped out. You can read an interesting, if ghoulish, account of Kuru soup making from 1949. Recipe included. Rumor has it New Guinea had to give
up the distinction, "home of Cannibal's Soup".
These two varieties of TSEs are inherited:
Gerstmann-Straussler Syndrome (GSS) makes up about 10-15% of confirmed cases. GSS is both inherited
and sporadic9 and occurs at about 2% of the rate of CJD.10 If a parent has GSS, there's a 50-50 chance children will develop
the disease.11
Fatal Familial Insomnia (FFI) - rare and has only been seen in about 9 families.
HOW DO YOU GET CJD?
There appear to be three ways to acquire CJD.
First, the disease can occur sporadically which means there is no known source and no one in the patient's family has the disease. CJD used to be thought of in connection with aging since most victims were over 45, but now the average age for CJD to strike is 26.5 and can show up in children. Sporadic CJD occurs about 1 per million per year.12
Second, the disease can be inherited which accounts for approximately 10 to 15% of CJD cases.
Third, CJD can be transmitted by infection. New information is being learned about transmission all the
time with more to know. People can be infected several ways:
 eating contaminated food eating contaminated food
Meat-on-the-bone has been off-menu in many European restaurants. McDonald's has also seen a large down turn in sales. This is not to say their foods are at risk, but people are being more cautious.
through medical and dental procedures using contaminated instruments
 (It is VERY hard to achieve complete sterilization. Prions survive
many standard chemical disinfectants and repeated sterilization
attempts using high temperatures. Doctors at the University of
Melbourne, showed that using heat sterilization - the standard
method of cleaning surgical instruments - actually makes it harder to destroy to prions. They now recommend using disposable instruments.13 This also raises serious questions about the safety of dental
visits.
(It is VERY hard to achieve complete sterilization. Prions survive
many standard chemical disinfectants and repeated sterilization
attempts using high temperatures. Doctors at the University of
Melbourne, showed that using heat sterilization - the standard
method of cleaning surgical instruments - actually makes it harder to destroy to prions. They now recommend using disposable instruments.13 This also raises serious questions about the safety of dental
visits.
|
- injection of human growth hormone from pituitaries of cadavers
By 1985, four children came down with CJD from taking human pituitary
growth hormones. Injection programs stopped immediately in most
countries, except France. They claimed their hormone-extraction
process was pure so the injections continued. By 1996, 100 cases
of CJD surfaced from using growth hormones; half were in France.14
In Australia, another human pituitary hormone, gonadotrophin,
was injected into 2100 infertile women between 1967 and 1985.
Five since have died of CJD. This treatment, too, was stopped
in 1985 when the same problem arose in the US.15 Most of these cases should have surfaced by now since incubation
takes 3 - 20 years.16
|
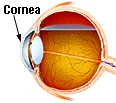
- receiving infected cornea transplants or having contam-
inated electrodes placed in the brain or through dural grafts17
Eyes, brains, spinal cords and tonsils are all areas of high prion concentration
|
|
|
- in utero - or from mother to fetus
This is a fairly new, frightening discovery.
|

|
|
MEET THE FIRST INFANT DIAGNOSED WITH CJD
What a heartbreaking start to a short little life. The implications
are extraordinary.
Amanda was born at the beginning of November, 1999 by cesarean
section. "Janet", a pseudonym given to the mother by the newspapers,
should have been on top of the world. Instead, before Amanda's
birth at 22, Janet showed many of the early symptoms of vCJD,
but was not diagnosed until later.

Doctors believe Amanda is the first child to contract the deadly
disease in utero. From the time the little girl was delivered,
Amanda had difficulty swallowing. At four months, she weighed
less than 10 pounds (4 kg), was blind and could only make yelping
noises. When doctors scanned Amanda's brain, they found the telltale
spongy lesions and she had never even tasted meat.
Despite extreme bouts of depression and illness, Amanda's mother was finally able to see her child four months after delivery. By this time, Janet had a staggering gait and she was barely able to control her drooling and body movements. Her speech and mannerisms had become childlike and due to a lack of coordination, Janet had to drink from a toddler's bottle. The visit was on a good day for Janet, but three months later, Janet was dead.
Continue
© Text and Graphics, 2001 Stan and Holly Deyo, except where otherwise
credited
|
